SCIENCE BITCH
Osoby wrażliwsze proszę o ominięcie tego postu, ponieważ może zawierać zdjęcia, na które nawet mnie ciężko patrzeć.
Co będą przedstawiać te zdjęcia? Najdziwniejsze i najobrzydliwsze mutacje genetyczne. Serio, to nie notka typu bardzo przyjemnie się czyta, suche żarciki.
Ale zanim dojdziemy do chorób trzeba liznąć trochę teorii. Jeśli nie zrozumiecie podstawowych pojęć to nie macie w sumie po co czytać notkę. Więc po kolei:
Podstawowe pytanie: Co to są mutacje?
Mutacje są to trwałe i nagłe zmiany w materiale genetycznym przekazywane z pokolenia na pokolenie.
Jest wiele czynników wywołujących mutację. Dzieli się je na :
- fizyczne (m.in. promieniowanie różnego rodzaju, wysoka temperatura)
- chemiczne (m.in. niektóre kwasy (np. kwas azotawy lub azotowy III - HNO₂), niektóre konserwanty (np. E950 tzw. Acesulfam K chilli inaczej sztuczny słodzik), barwniki np. oranż akrylowy, niektóre składniki dymu tytoniowego (benzen), gaz bojowy - iperyt)
- biologiczne (m.in. niektóre wirusy (np. wirus opryszczki) i mikroorganizmy)
Są to mutageny zewnętrzne. Niektóre z nich są kancerogenne (wywołują nowotwór) w tym:
- promieniowanie UV
- podwyższona temperatura
- kwas azotowy III
- iperyt
- oranż akrylowy
- benzen
- dioksyny
Jednak nie tylko one przyczyniają się do mutacji. Również może to być błąd podczas replikacji DNA. Polimeraza (enzym, który odczytuje nić matrycową i na jej wzór tworzy nową, komplementarną (chilli w DNA adeninie odpowiada tyrozyna a guaninie cytozyna i aby nić była komplementarna to musi się zgadzać) przecież też może się pomylić.
Ogólnie w tej notce mam zamiar skupić się na mutacjach genowych. I tutaj głównie będą nam potrzebne takie terminy:
nukleotyd - jednostka, która składa się na budowę DNA. Każdy nukleotyd, składa się z cukru (deoksyrybozy), zasady azotowej i reszty kwasu fosforowego.
delecja - utrata jednej lub większej liczby par nukleotydów z DNA
inwersja - zmiana zwykłego układu nukleotydów lub chromosomów na odwrotny
duplikacja - podwojenie
translokacja - wymiana odcinków chromosomów między chromosomami niehomologicznymi (zupełnie do siebie niepodobnymi) lub przeniesieniu fragmentu chromosomu na inny.
Musimy też poznać podział genomopatii (mutacji genowych). Dzielą się one na:
- eupoidalne (poliploidalne) czyli zwielokrotnienie podstawowej liczby chromosomów o jeden lub kilka kompletów. Wiedząc, że n to gametyczna liczba chromosomów, chilli liczba chromosomów znajdująca się w gamecie osobnika możemy wyróżnić haplonty (n), diplonty (2n), triplonty inaczej triploidy (3n), tetraploidy (4n), pentaploidy (5n), heksaploidy (6n) itd.
- aneuploidalne czyli zwiększenie lub zwiększenie podstawowej chromosomów o jeden lub dwa chromosomy.
- trisomie 2n + 1, gdzie 2n to podstawowa liczba chromosomów u człowieka ofc (np. Zespół Downa)
- tetrasomie 2n + 2
- monosomie 2n - 1
- nullisomie 2n - 2
Podział jest identyczny jednak różnica jest znaczna, ponieważ tetrasomia, monosomia i nullisomia jako mutacja autosomalna jest letalna czyli śmiertelna. Nikt nie wie jak dokładnie wygląda osoba z taką mutacją, bo wszystkie umierają jako płód. Oczywiście można sprawdzić ryzyko wystąpienia tych mutacji badaniami prenatalnymi.
W przypadku mutacji allosomalnych letalna jest tylko nullisomia.Dlaczego? To bardzo proste.
W przypadku mutacji autosomalnych zmienia się liczba autosomów a w przypadku allosomalnych liczba allosomów (chromosomów płci). Właśnie rodzaj chromosomów, na których zachodzi mutacja warunkuje to, czy mutacja będzie letalna czy nie. Dodatkowy chromosom X czy Y nie wpływa w tak znaczący sposób na organizm ludzki jak dodatkowy chromosom w np. 21 parze chromosomów (chilli inaczej mówiąc Zespół Downa).
Dzisiaj opowiem wam o najobrzydliwszych i najdziwniejszych mutacjach czyli w kilku zdaniach opowiem wam jak wiele może zmienić jeden chromosom
. No i też najciekawszych bądź co bądź.
Teraz wrażliwe osoby mogą już sobie iść, bo będą zdjęcia.
Zespół Patau'a czyli dzieci - cyklopy
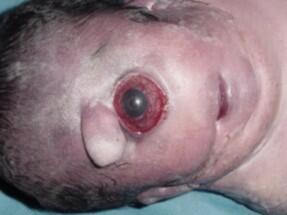
Proszę mi uwierzyć, że wybrałam jedno z najmniej obrzydliwych zdjęć tej choroby. Jak mi nie wierzycie to wygooglujcie (chociaż nie polecam).
Ale od początku: Zespół Patau'a jest trisiomią 12 chromosomu. Charakteryzuje go (oprócz oczywistego) duże ryzyko poronienia, niska masa urodzeniowa, obniżenie napięcia mięśniowego
ubytek skóry skalpu, holoprozencefalia (niedokonany podział przodomózgowia), ciężkie wady narządu wzroku (włączając w to cyklopie - zamiast dwóch gałek ocznych jest jedna), nieprawidłowo wykształcony nos, wargi, podniebienia, liczne wady małżowin usznych, np. niskie osadzenie), anomalie kończyn (np. dodatkowe palce u rąk lub u nóg, ustawienie palców w kształcie "kurka od strzelby", wydatna pięta. Ponadto można zdiagnozować wady serca, nerek, wady rozwojowe mózgowia i cewy nerwowej, u chłopców wnętrostwo, a u dziewczynek wady rozwojowe macicy.
To ja wiele z tych objawów wystąpi na raz zależy od tego, ile dodatkowego chromosomu powstało. W większości dotyka dziewczynek głównie dlatego, że płody płci męskiej nie dożywają do porodu. Około 10% dzieci z tym zespołem umiera w ciągu pierwszego półrocza życia, ponad 90% nie dożywa pierwszego roku życia a maksymalnie 5% dożywa trzech lat.
Zespół Patau można rozpoznać już w drugim trymestrze ciąży. Wówczas podczas badania USG można dostrzec m.in. opóźnienie rozwoju fizycznego czy małogłowie. Więc teoretycznie można uchronić dziecko przed miesiącami cierpień. Wystarczy tylko iść na badania prenatalne.
Zespół Edwardsa

Zespół Edwardsa to choroba genetyczna polegająca na pojawieniu się dodatkowego, trzeciego, chromosomu 18. w DNA (trisomia chromosomu 18. pary). Zdarza się raz na 5 000 (według innych danych raz na 3 500) urodzeń zwłaszcza u dziewczynek - cztery razy częściej niż u chłopców. Ta choroba genetyczna niemal w 95 % prowadzi do samoistnego poronienia w pierwszym trymestrze ciąży. Większość dzieci urodzonych żyje najwyżej dwa miesiące, ale od 5 do 10 % żyje więcej niż rok. To zależy od ciężkości i ilości wad, które wywołuje choroba.
Charakterystyczne objawy to m.in. niedorozwój umysłowy,wady serca, nerek, przewodu pokarmowego, nieprawidłowo zbudowana czaszka, rozszczepy wargi, podniebienia, niska waga płodu oraz nieprawidłowy rozwój kośćca - karku i szyi oraz kostki nosowej i klatki piersiowej, a także wady ośrodkowego układu nerwowego.
Można go wykryć badaniami prenatalnymi.
Zespół Wolfa-Hirschhorna

Przyczyną choroby jest mikrodelecja fragmentu jednego z pary chromosomów 4, czyli utrata odcinka DNA. W jednej z par chromosomu 4 brakuje fragmentu krótkiego ramienia, który wpływa na powstanie wad wrodzonych. Utrata części składników materiału genetycznego następuje jeszcze na etapie formowania się genotypu (indywidualnego zestawu genów). Wielkość ubytku nie ma wpływu na nasilenie objawów chorobowych, ponieważ każda, nawet najmniejsza mikrodelecja prowadzi do pojawienia się choroby. Typowa dla tego zespołu jest niska masa urodzeniowa, hipotonia (obniżenie napięcia mięśniowego), małogłowie (nienaturalnie mały rozmiar czaszki), niepełnosprawność intelektualna.Charakterystyczna jest również dysmorfia twarzy, która wyglądem przypomina hełm grecki (tzn. czoło jest wysokie i wydatne) i nieprawidłowo mała żuchwa. Zauważalne są również szeroko rozstawione oczy (tzw. hiperteloryzm oczny), opadające powieki, szeroki dziobiasty nos, nisko osadzone uszy, duże małżowiny uszne. W niektórych przypadkach widoczny może być również rozszczep warg i podniebienia. O genetycznych nieprawidłowościach mogą świadczyć także wady budowy narządów płciowych zewnętrznych i wewnętrznych (u chłopców np. spodziectwo, u dziewczynek np. niedorozwój pochwy, macicy), a także wady serca, układu kostnego, układu pokarmowego, wzorku. Osłabiony jest system odpornościowy chorego.
Diagnozę można postawić nie tylko po narodzinach dziecka, lecz także we wczesnym etapie rozwoju płodu. W tym celu należy wykonać badania prenatalne, takie jak nieinwazyjne badanie USG oraz amniocentezę (pobranie niewielkiej próbki płynu owodniowego), biopsję kosmówki czy przezskórne pobranie krwi pępowinowej (są to badania inwazyjne, w związku z tym niosą za sobą ryzyko powikłań). W najlepszym przypadku chory dożywa wieku dorosłego. Nie da się go uleczyć, można jedynie złagodzić objawy.
Progeria: przedwczesna starość.
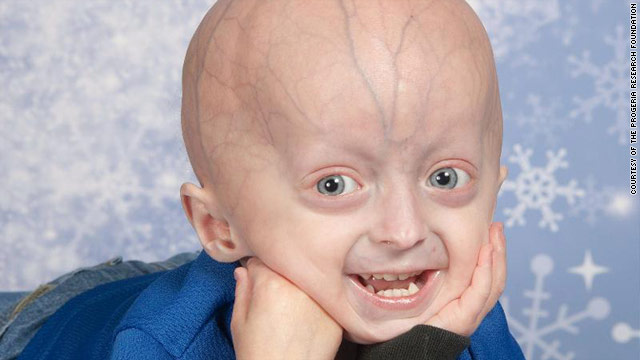
Ma dwie zasadnicze odmiany: progeria u dzieci występuje w postaci syndromu Hutchinsona-Gilforda, a progeria u dorosłych ma postać zespołu Wernera. Różnica między nimi polega na momencie w życiu człowieka, w jakim się ujawniają.
Syndrom progerii Hutchinsona-Gilforda, zwany tak od nazwisk lekarzy, którzy prowadzili badania nad tą dziwną przypadłością, objawia się u małych dzieci, najczęściej gdy skończą rok. Dzieci obciążone progerią w momencie urodzenia mają zazwyczaj prawidłową masę ciała i w pierwszych dwunastu miesiącach życia – poza ewentualnymi nieustępującymi zmianami (wypryski i zaczerwienienia) na skórze, choroba przebiega bezobjawowo. Jej istnienie rodzice zaczynają podejrzewać, gdy skóra dziecka wiotczeje, tworzą się na niej pierwsze zmarszczki, wypadają włosy, dziecko ząbkuje z opóźnieniem, nie rośnie i nie przybiera prawidłowo na wadze. To dzieje się najczęściej już w drugim roku życia. W kolejnych latach następują rozleglejsze zmiany: dziecko jest niskie i szczupłe, ma jednak nieproporcjonalnie duża głowę z cofniętą, niedorozwiniętą żuchwą, w której często nie mieszczą się zęby, wyraźne żyły na skórze głowy, wyłupiaste oczy. Klatka piersiowa przybiera nieregularny kształt, obojczyki są krótkie, biodra koślawe. Na chudych kończynach wyraźnie wystają stawy. Skóra jest cienka i sucha, mnożą się na niej zmarszczki i plamy charakterystyczne dla starszego wieku. Zdarza się, że młodzi ludzie cierpią na osteoporozę zlokalizowaną w nietypowych miejscach – najczęściej w kościach długich nóg. Złogi wapnia lokują się także w tkankach miękkich, a suchość skóry sprzyja powstawaniu wrzodów w okolicach łokci, stawów skokowych, na stopach. Dziecko nie dojrzewa płciowo, ma wysoki, piskliwy głos. Jednak jego rozwój umysłowy przebiega bez zakłóceń, a czasami obserwuje się nawet ponadprzeciętne uzdolnienia.
Progeria objawiająca się u ludzi dorosłych nazywana jest zespołem Wernera. Pierwsze jej objawy dostrzegalne są w okresie dojrzewania, a wszystkie ujawniają się u dwudziestokilkulatków. I choć naukowcy nie ustalili jeszcze, czy schorzenie to naprawdę przyspiesza normalne procesy starzenia się, czy tylko je przypomina, to efekt jest taki, że człowiek dotknięty tą zespołem Wernera w ciągu jednego roku starzeje się o 7-8 lat. Umiera zatem szybko.
Progeria to nie tylko objawia się szybkim starzeniem, ale towarzyszą jej wszystkie choroby 'starych ludzi' np. artretyzm, reumatyzm itd.
Jest jeszcze masa takich chorób, ale myślę, że na początek tyle wystarczy. Znając życie, świat i mnie jeśli się wam spodoba to wrócę do tematu. Na razie wracam do nadrabiania notek i ogólnie pisania tutaj, bo bardzo zaniedbałam blog. So jeśli przetrwaliście do końca napiszcie komentarz, anonimowe są włączone a będzie mi bardzo miło zobaczyć co o tym myślicie.
Cya,
Neff
Jezu, ja chyba nie opanowałam dodawania tu komentarzy, whatever.
OdpowiedzUsuńWięc tak: podziwiam cię za research, bo widać że porządnie się przygotowałaś, więc też za rzetelność. Sposób w który piszesz jest przejrzysty nawet dla mniej lotnego umysłu (czyt. mnie), więc też gratki. Temat bardzo dobry, ciekawy, chociaż przykry. W sumie to trochę okrutne i chyba dla części z tych dzieci lepiej byłoby się nie urodzić niż żyć w męczarniach (przepraszam za poruszenie tego tematu, ale to się strasznie ciśnie na usta).
Mam jedną uwagę: Zespół Pataua mogłaś dać na sam koniec, bo jest chyba najbardziej drastyczny i mimo wszystko zdjęcie zaskakuje okrutnością mutacji, mam nadzieję że rozumiesz (bo chyba piszę od rzeczy).
Fajnie byłoby, gdybyś pisała teraz systematycznie.
Częściowo w notce chciałam pokazać co można wykryć badaniami prenatalnymi i czemu zapobiec, bo każdą z tych chorób czy też możliwość jej wystąpienia da się zbadać na początku ciąży a nawet wcześniej. Zespół Patau'a nie jest najbardziej drastyczny, są o wiele gorsze, ale to by było zbyt drastyczne nawet jak na mnie. Zdjęcie wybrałam też takie w miarę, bo są o wiele gorsze np. gdy dziecko ma dwie całki oczne i nadal jest cyklopem a do tego kilka rozszczepów twarzy. W planach miałam napisać więcej dlatego stwierdziłam, że jak kogoś nie zniechęci on na samym początku to doczyta już resztę.
UsuńNotkę w większości pisałam z głowy, te informacje przyswoiłam sobie dawno, dawno temu. Jedyne co, to szukałam zdjęć i sprawdzałam we własnych notatkach jak nie byłam pewna. Mogłam napisać wszystko naprawdę skomplikowanym typowo biologicznym językiem, ale to by nie miało sensu jeśli ktoś, kto nie ma głowy do przedmiotów ścisłych próbowałby to przeczytać, więc ograniczyłam się do minimum.
Jeśli większość z tych informacji miałaś już przyswojone, to ja naprawdę Cię podziwiam. Zespół Patau'a jest chyba najbardziej drastyczny z tych, które przedstawiłaś - o to mi chodziło.
UsuńMyślę, że dobrze zrobiłaś, nie pisząc tego w tak skomplikowany sposób, bo dla kogoś nieobeznanego, nawet jeśli ma ten ścisły umysł, mogłoby to być ciężkie do czytania.
Chętnie przeczytałabym drugą część tej notki jeśli kiedykolwiek powstanie (zapomniałam to wcześniej napisać, taktak).
Mała korekta - zespół Pataua to trisomia nie 12, lecz 13 chromosomu.
OdpowiedzUsuńzespol Pataua to trisomia 13 chromosoumu a nie 12 :)
OdpowiedzUsuń